
 Co-responsables : Denis Furling & Geneviève Gourdon
Co-responsables : Denis Furling & Geneviève Gourdon
La thématique principale de l’équipe est centrée sur la Dystrophie Myotonique (DM), une des maladies neuromusculaires les plus fréquentes chez l’adulte, et plus particulièrement sur la DM de type 1 (DM1) également appelée maladie de Steinert.
La DM1 est caractérisée par une faiblesse et une atrophie musculaire progressive, une myotonie, des défauts de conduction cardiaque, une cataracte, des troubles endocriniens et gastro-intestinaux ainsi que des atteintes neurologiques. On distingue cinq formes de cette maladie multisystémique incluant les formes tardive, adulte, juvénile, infante et congénitale. Actuellement, il n’y a pas de traitement pour cette maladie génétique mais diverses approches thérapeutiques sont en développement.
La DM1 est une maladie autosomique dominante causée par une expansion de triplets CTGn (n>40) localisée dans la région 3’ non codante du gène DMPK. Le caractère instable de cette expansion de séquences CTG répétées se manifeste au niveau somatique tout au long de la vie du patient mais également entre générations successives, et représente la base moléculaire du phénomène d’anticipation observé dans cette maladie. La DM1 est une maladie à gain-de-fonction d’ARN. En effet, les transcrits DMPK mutés contenant des répétitions CUG pathologiques (ARN-CUGexp) sous retenus dans le noyau des cellules sous forme d’agrégats riboprotéiques (foci) qui vont notamment séquestrer les protéines de liaison aux ARNs de la famille MBNL impliquées dans le processus de maturation des ARNs. Ainsi, la perte fonctionnelle de MBNL conduit à des défauts d’épissage alternatif de certains ARN pré-messagers qui ont été associés à des symptômes comme ceux du CLCN1 à la myotonie, de BIN1 à la faiblesse musculaire, de DMD à l’altération de l’architecture des fibres musculaires et de SCN5A à des défauts de conduction cardiaque et du rythme. Cependant des mécanismes additionnels sont impliqués dans les processus physiopathologiques complexes de cette maladie qui affecte divers types cellulaires et de nombreux tissus.
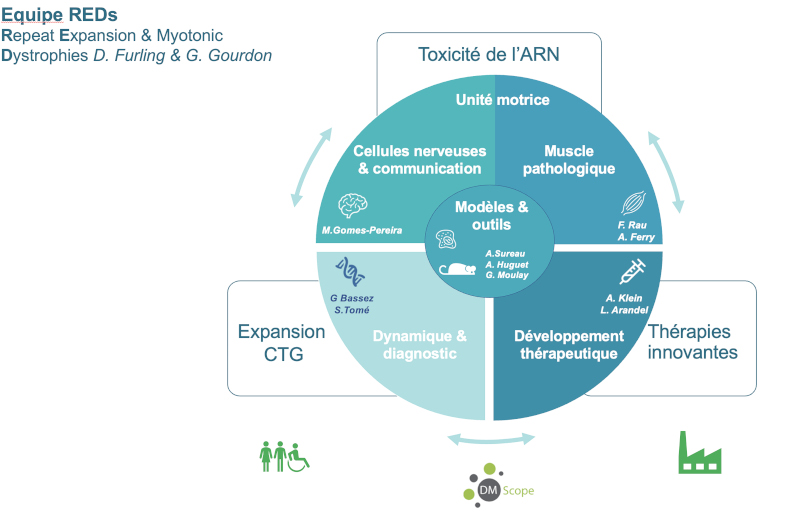
L’équipe REDs a été créée en 2019 suite à la fusion des équipes de Geneviève Gourdon et Denis Furling. Elle inclut également le groupe de Guillaume Bassez qui coordonne le registre national de dystrophie myotonique DM-Scope. L’objectif de cette nouvelle équipe est de synergiser les efforts afin d’accélérer la recherche translationnelle pour cette maladie neuromusculaire et de faire émerger des alternatives thérapeutiques pour les patients. Ainsi les compétences de l’équipe visent à faire une recherche intégrée autour de la DM1 allant de la mutation à la compréhension des mécanismes physiopathologiques à l’aide de modèles cellulaires et animaux, au développement et l’évaluation d’approches thérapeutiques innovantes et enfin, à la mise en place d’essais pré-cliniques et cliniques pour cette maladie neuromusculaire.