 Team leader: Dr Teresinha Evangelista, MD
Team leader: Dr Teresinha Evangelista, MD
Head of the histology platform: Nadine Vailhen, PhD
The Risler Pavilion Laboratory is dedicated in particular to the morphological identification and characterisation of genetic neuromuscular diseases in the adult and child. The Morphological Unit is part of the European Reference Network for Rare Neuromuscular Diseases (ERN EURO-NMD) and the French Reference Centre for Neuromuscular Diseases North / East / Ile-de-France
This laboratory is recognised for the scientific value of its work, its experience in human muscle biopsy analysis and its mastery of morphological techniques. Several fundamental and complementary activities are also carried out:
- Histo-enzymological analyses of frozen muscle sections
- Immunocytochemical analyses using specific antibodies directed against different muscle cell proteins : (dystrophin, sarcoglycans, dystroglycans, dysferlin, caveolin, collagen, desmin, myotilin, myosin etc.)
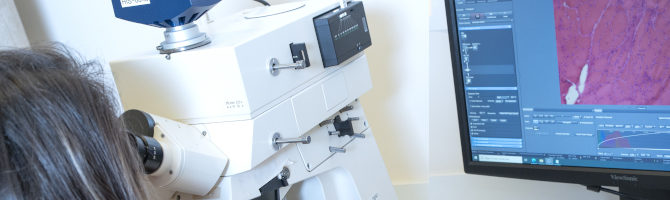
- Ultrastructural analyses by electron microscopy
- Western blot analysis
- MND diagnosis
- Centre for reference for neuromuscular disease diagnosis
- Designing or participating in research programmes necessitating this expertise
- Training

Team members
Dr Teresinha Evangelista, MD, Team leader, Hospital Practitioner, researcher
Dr Béatrice Labella, MD, Neurologist specialised in neuromuscular pathology
Nadine Vailhen, PhD, Head of histopathology platform
Favienne Levy-Borsato, Secretary
Emmanuelle Lacene, Clinical Research Assistant
Mai Thao Bui, Research Technician
Guy Brochier, PhD, Research Technician Assistant
Angéline Madelaine, Laboratory Technician
Anaïs Chanut, Laboratory Technician
Clémence Labasse, Research Technician
Maud Beuvin, Laboratory Assistant
Dr Norma B. Romero, MD, PhD, Volunteer Consultant
Contact
01 42 16 22 42
01 42 16 22 43
Diagnosis of NMD
Reference centre for diagnosis of NMD
Participation in research projects
Training
Last publications
Diagnosis of NMD
We analyse muscle biopsies in our laboratory, with the precise aim of diagnosing muscle pathologies. Some muscle samples from the laboratory’s collection are re-analysed using new tools resulting from advanced research methods.
This activity is very directly linked with the concrete needs of patients.
Requests come either from the Pitié-Salpêtrière Hospital Group: the Neuromuscular Pathology consultation of the Institute of Myology at the Salpêtrière (Prof Bertrand Fontaine), the Internal Medicine department of Dr Olivier Benveniste, or from hospitals outside the Pitié-Salpêtrière Group (Necker-Children’s, Robert Debré, Val de Grâce, Institute of Childcare and Rothschild in Paris, Raymond Poincaré in Garches, Central Hospitals of Poissy, Montreuil etc).
Reference centre for diagnosis of NMD
In our capacity as a Centre of Expertise of Neuromuscular Pathology, we are asked to respond to requests from other centres in France and abroad.
We are frequently approached to:
- give a second opinion about examinations already carried out elsewhere ; we regularly receive biopsy sections in cases of diagnosis difficulties (about sixty cases a year).
- complete the analyses of muscle samples taken by other laboratories in Paris or the provinces. These samples are analysed using specialist techniques, not available in all laboratories.
- help different French and foreign laboratories (by taking samples for research).
Participation in research projects
The aim of this work is to contribute to the characterisation of new or as yet unidentified neuromuscular diseases (in collaboration with : M. Bitoun, G. Bonne, D. Hantaï, A. Ferreiro, F. Leturcq, P. Richard, J. Lunardi, N. Monnier, J. Laporte, V. Biancalana, A. Oldfors, F. Muntoni, N. Clarke, N. Laing, C.Wallgren-Pettersson, AL Taratuto etc) . The goal of this work is to contribute at the characterisation of new or not yet identified neuromuscular diseases (in collaborations).
To be mentioned here:
- The first protocol of gene therapy done in humans. This phase I essay of clinical research, without any direct benefit for the patients, has been performed using a plasmid with total cDNA of dystrophin in patients with disease of Duchenne or Becker. The principal investigators were Pr Michel Fardeau (Institut de myologie) and Pr Serge Herson (Internal medicine CHUPS). This essay has been performed in collaboration with the Transgene collaboration.
- The immunocytochemical, histo-enzymological and ultrastructural analyses of abnormalities associated with congenital myopathies, in particular core myopathies (Central Core Disease (CCD), Multi-minicore), correlated with molecular studies, allows to bring precisions at the clinical entities which are not yet well defined or differentiated. In the domain of congenital myopathies with cores, we have also contributed to the clinical and morphological characterisation of patients with CCD linked or not-linked with the gene RYR1.
- In the domain of congenital myopathies, we have also contributed at the study of nemalin myopathies (NM) with the identification of one of the first children with a severe form of NM, which was associated at a mutation stop of the gene ACTA1 and at the study of myopathies with congenital fiber type disproportion (CFTD) linked at the gene TPM3.
- Thanks to the immunocytochemical study with antibodies against alpha- and beta-dystroglycan, collagen VI and perlecan, which we have systematically introduced in our laboratory, it has been possible to perform a reclassification of multiple muscular dystrophies of at that time unknown origin. In this domain, we have contributed to the identification of congenital muscular dystrophy associated at collagen 6, as well as to the study of muscular dystrophies with deficiency in alpha-dystroglycan, linked or not-linked at the gene FKRP.
- A first clinical and morphological analysis of a large series of patients with a congenital myopathy of the centronuclear type (CNM) has been performed. This work has contributed in an important way to linkage analyses on the autosomal dominant form of CNM and to the identification of the causal gene DNM2 that encodes Dynamine 2. Next, we have described the selectivity of the muscular affection in patients with a DNM2 mutation by a clinical and muscle imaging study. Later, we have identified the severe clinical and sporadic forms associated with DNM2. Next, we concentrated our interest on the histopathological analysis of patients in whom a mutation in DNM2 had been excluded. These patients make up a heterogeneous group with as common characteristic a variable number of fibres with internalized nuclei. This work has led to a classification in several subgroups of patients based on the morphological aspects observed at the muscular biopsy, as well as the mode of transmission and the clinical presentation.
- We have also identified a group of CNM patients that presented a curious morphological feature characterized by the presence of a variable number of fibers « en collerettes » or “dark necklace” fibres (DNF). These dark necklaces are situated a few micrometers of the sarcolemma and have been observed in the two types of fibres. The dark necklaces are colored by HE, GT, PAS, NADH-TR, SDH and COX, but they are not stained by myofibrillar ATPase. Some nuclei are aligned with the dark necklace. The ultrastructural analysis showed that the dark necklace consists of myofibrils with a small diameter, which are surrounded by mitochondria and sarcotubular structures. The immunohistochemical analysis has shown an intense staining with the anti-SERCA1 and SERCA2 antibodies, bot not with the other proteins of the sarcoplasmic reticulum (calsequestrin, the ryanodin receptor, triadin), and the T-tubule (dihydropyridin receptor-alpha1subunit). We have observed a moderate reaction with the anti-desmin and alphaB-crystallin antibodies, but less with anti-myotilin. Clinically, it is sporadic cases, with symptoms that started in the first decennium of their lives, with a proximal weakness of the lower limbs and a slowly progressive evolution. The genetic analysis has permetted to exclude mutations in the genes DNM2, hJumpy and BIN. The analysis of the gene MTM is currently performed. The mechanism of this defect in the organisation of the sarcoplasm still has to be elucidated.
- We are studying the clinical, immunohistochemical and ultrastructural characteristics in patients with myofibrillar myopathy (MFM). The ultrastructural findings in 19 patients with different genetically-proven MFMs (9 desmin, 5 alpha-B-crystallin, 3 ZASP, 2 myotilin) were analyzed. In one patient with a ZASP mutation, we additionally performed an immuno-electron microscopic (EM) study, using antibodies against desmin, alpha-B-crystallin, ZASP and myotilin. The ultrastructural findings in desminopathies and alpha-B-crystallinopathies were very similar and consisted of electrondense granulofilamentous accumulations and sandwich formations. They differed in the presence of early apoptotic nuclear changes in alpha-B-crystallinopathies. ZASPopathies were characterized by filamentous bundles (labeled with the myotilin antibody on immunoEM), and floccular accumulations of thin filamentous material. Tubulofilamentous inclusions in sarcoplasm and myonuclei in combination with filamentous bundles were characteristic for myotilinopathies. We conclude from this study that MFMs ultrastructural findings can direct diagnostic efforts towards the causal gene mutated, and that EM should be included in the diagnostic workup of MFMs. The immunohistochemical study involves 15 patients with a genetically-proven MFM (7 desmin, 3 alpha-B-crystallin, 3 ZASP, 2 myotilin) in whom we used 23 antibodies including several novel antibodies against distinct Z-disc and M-line proteins. This study is performed in collaboration with Dieter O. Fürst and Peter F.M. van der Ven of the Institute for Cell Biology, Department of Molecular Cell Biology, University of Bonn, Bonn (Germany) and with Rudolf Kley of the Neurologische Univ.-Klinik Bergmannsheil, Ruhr-Universität Bochum, Bochum (Germany). The results of this study are currently being analyzed.
Training
Teams working on the study of muscular diseases have expressed an interest in following up or improving their training in muscular biopsy analysis (example: Study group Meetings in Myology organised at the Institute of Myology etc).
For several years now, we have received physicians for training courses in the diagnosis of muscular diseases and/or postdoctoral trainings, and technicians for training in sampling techniques and the processing of biopsies. Last years, we received Michele Roccella (Palerme), Pierre-Yves Jeannet, (Lausanne), Haffed Haddad (Tunis), Simona Gambelli (Sienne), Muriel Herasse (Paris), Fabiana Lubieniecki (Buenos Aires), Soledad Monges (Buenos Aires) et Javier Linzoain (Córdoba). In 2008, we will welcome Felipe Andreinolo, neuropathologist of Brasil (Sao Paulo) and Jorge Bevilacqua, neurologist of Chili (Santiago)
The Risler Pavilion now has a specially adapted and equipped meeting room for up to 15 people in order to bring together physicians and scientists with an interest in Neuromuscular Pathology. Several regular meetings are held there:
- Meetings to compare genetic/morphologic/pathological/clinical results (monday) with the regular participation of Dr F. Leturcq, Dr S. Quijano, Dr P. Laforet, Dr G. Bonne, Dr R. Ben Yaou, Dr P. Richard, Dr A. Behin, Dr T. Stojkovic, Dr T. Evangelista, Dr C. Métay.
- Patient record discussion meetings (Thursday), between the physicians of the consultation of the Institute of Myology (Dr P. Laforet, Dr A. Behin, Dr T. Stojkovic, Dr T. Evangelista, Dr G. Bassez) and the physicians of the Internal Medicine department (Dr O. Benveniste).
- Metabolic diseases meetings (Friday) with the participation of the biologists of the laboratory of Prof B.Hainque.
Although the laboratory of the Risler Pavilion is integrated within the hospital, it collaborates with other inter- and/or extra-hospital partners : the neuromuscular pathology consultation of the Babinski Building, the Raymond Escourolle Neuropathology Laboratory, the Tissue Bank for Research, the Internal Medicine department, the Cardiology department, the Immunology department, the Federation of Neurology and the CNRS and Inserm Research Units.
Last publications
- Autosomal dominant in cis D4Z4 repeat array duplication alleles in facioscapulohumeral dystrophy. Lemmers RJLF, Butterfield R, van der Vliet PJ, de Bleecker JL, van der Pol L, Dunn DM, Erasmus CE, D’Hooghe M, Verhoeven K, Balog J, Bigot A, van Engelen B, Statland J, Bugiardini E, van der Stoep N, Evangelista T, Marini-Bettolo C, van den Bergh P, Tawil R, Voermans NC, Vissing J, Weiss RB, van der Maarel SM. Brain. 2024 Feb 1;147(2):414-426. doi: 10.1093/brain/awad312. PMID: 37703328 Free PMC article.
- Current management of primary mitochondrial disorders in EU countries: the European Reference Networks survey. Mancuso M, Lopriore P, Lamperti C, Klopstock T, Rahman S, Licchetta L, Kornblum C, Wortmann SB, Dollfus H, Papadopoulou MT, Arzimanoglou A, Scarpa M, Graessner H, Evangelista T. J Neurol. 2024 Feb;271(2):835-840. doi: 10.1007/s00415-023-12017-1. Epub 2023 Oct 13. PMID: 37831128 Free PMC article.
- EURO-NMD registry: federated FAIR infrastructure, innovative technologies and concepts of a patient-centred registry for rare neuromuscular disorders. Atalaia A, Wandrei D, Lalout N, Thompson R, Tassoni A, ‘t Hoen PAC, Athanasiou D, Baker SA, Sakellariou P, Paliouras G, D’Angelo C, Horvath R, Mancuso M, van der Beek N, Kornblum C, Kirschner J, Pareyson D, Bassez G, Blacas L, Jacoupy M, Eng C, Lamy F, Plançon JP, Haberlova J, Brusse E, Hoeijmakers JGJ, de Visser M, Claeys KG, Paradas C, Toscano A, Silani V, Gyenge M, Reviers E, Hamroun D, Vroom E, Wilkinson MD, Lochmuller H, Evangelista T. Orphanet J Rare Dis. 2024 Feb 14;19(1):66. doi: 10.1186/s13023-024-03059-3. PMID: 38355534 Free PMC article. Review.
- Phenotype variability and natural history of X-linked myopathy with excessive autophagy. Fernández-Eulate G, Alfieri G, Spinazzi M, Ackermann-Bonan I, Duval F, Solé G, Caillon F, Mercier S, Pereon Y, Magot A, Pegat A, Salort-Campana E, Chabrol B, Gorokhova S, Krahn M, Biancalana V, Evangelista T, Behin A, Metay C, Stojkovic T. J Neurol. 2024 Jul;271(7):4008-4018. doi: 10.1007/s00415-024-12298-0. Epub 2024 Mar 22. PMID: 38517523
- Management of seizures in patients with primary mitochondrial diseases: consensus statement from the InterERNs Mitochondrial Working Group. Mancuso M, Papadopoulou MT, Ng YS, Ardissone A, Bellusci M, Bertini E, Di Vito L, Evangelista T, Fons C, Hikmat O, Horvath R, Klopstock T, Kornblum C, Lamperti C, Licchetta L, Molnar MJ, Varhaug KN, O’Callaghan M, Pressler RM, Schiff M, Servidei S, Szabo N, Gorman GS, Cross JH, Rahman S. Eur J Neurol. 2024 Jul;31(7):e16275. doi: 10.1111/ene.16275. Epub 2024 Apr 4. PMID: 38576261 Free PMC article.
- IMPatienT: An Integrated Web Application to Digitize, Process and Explore Multimodal PATIENt daTa. Meyer C, Romero NB, Evangelista T, Cadot B, Laporte J, Jeannin-Girardon A, Collet P, Ayadi A, Chennen K, Poch O. J Neuromuscul Dis. 2024;11(4):855-870. doi: 10.3233/JND-230085. PMID: 38701156 Free PMC article.
- Model matchmaking via the Solve-RD Rare Disease Models & Mechanisms Network (RDMM-Europe). Ellwanger K, Brill JA, de Boer E, Efthymiou S, Elgersma Y, Icmat M, Lecoquierre F, Lobato AG, Morleo M, Ori M, Schaffer AE, Vitobello A, Wells S, Yalcin B, Zhai RG, Sturm M, Zurek B, Graessner H, Bermejo-Sánchez E, Evangelista T, Hoogerbrugge N, Nigro V, Schüle R, Verloes A, Brunner H, Campeau PM, Lasko P, Riess O. Lab Anim (NY). 2024 Jul;53(7):161-165. doi: 10.1038/s41684-024-01395-2. PMID: 38914824 Free PMC article.
- Rapidly progressive myopathy: unveiling light chain amyloidosis as an initial manifestation of multiple myeloma: a case report and literature review. Kaminskiene P, Stojkovic T, Roos-Weil D, Reimbold P, Chanut A, Lacene E, Evangelista T. Neuromuscul Disord. 2024 Aug;41:51-55. doi: 10.1016/j.nmd.2024.06.002. Epub 2024 Jun 6. PMID: 38925009 Free article. Review.
- French National Protocol for diagnosis and care of facioscapulohumeral muscular dystrophy (FSHD). Attarian S, Beloribi-Djefaflia S, Bernard R, Nguyen K, Cances C, Gavazza C, Echaniz-Laguna A, Espil C, Evangelista T, Feasson L, Audic F, Zagorda B, Milhe De Bovis V, Stojkovic T, Sole G, Salort-Campana E, Sacconi S. J Neurol. 2024 Sep;271(9):5778-5803. doi: 10.1007/s00415-024-12538-3. Epub 2024 Jul 2. PMID: 38955828 Review.
- Defining the landscape of TIA1 and SQSTM1 digenic myopathy. Panos-Basterra P, Theuriet J, Nadaj-Pakleza A, Magot A, Lannes B, Marcorelles P, Behin A, Masingue M, Caillon F, Malek Y, Fenouil T, Bas J, Menassa R, Michel-Calemard L, Streichenberger N, Simon JP, Bouhour F, Evangelista T, Métay C, Pegat A, Stojkovic T, Fernández-Eulate G. Neuromuscul Disord. 2024 Sep;42:43-52. doi: 10.1016/j.nmd.2024.07.008. Epub 2024 Jul 24. PMID: 39142003
- Dysregulation of muscle cholesterol transport in amyotrophic lateral sclerosis. Sapaly D, Cheguillaume F, Weill L, Clerc Z, Biondi O, Bendris S, Buon C, Slika R, Piller E, Sundaram VK, da Silva Ramos A, Amador MDM, Lenglet T, Debs R, Le Forestier N, Pradat PF, Salachas F, Lacomblez L, Hesters A, Borderie D, Devos D, Desnuelle C, Rolland AS, Periou B, Vasseur S, Chapart M, Le Ber I, Fauret-Amsellem AL, Millecamps S, Maisonobe T, Leonard-Louis S, Behin A, Authier FJ, Evangelista T, Charbonnier F, Bruneteau G. Brain. 2025 Mar 6;148(3):788-802. doi: 10.1093/brain/awae270. PMID: 39197036 Free PMC article.
- An interconnected data infrastructure to support large-scale rare disease research. Johansson LF, Laurie S, Spalding D, Gibson S, Ruvolo D, Thomas C, Piscia D, de Andrade F, Been G, Bijlsma M, Brunner H, Cimerman S, Dizjikan FY, Ellwanger K, Fernandez M, Freeberg M, van de Geijn GJ, Kanninga R, Maddi V, Mehtarizadeh M, Neerincx P, Ossowski S, Rath A, Roelofs-Prins D, Stok-Benjamins M, van der Velde KJ, Veal C, van der Vries G, Wadsley M, Warren G, Zurek B, Keane T, Graessner H, Beltran S, Swertz MA, Brookes AJ; Solve-RD consortium. Gigascience. 2024 Jan 2;13:giae058. doi: 10.1093/gigascience/giae058. PMID: 39302238 Free PMC article.
- Relevance of muscle biopsies in the neonatal and early infantile period: a 52 years retrospective study in the gene-sequencing era. Bui MT, Fernández-Eulate G, Evangelista T, Lacène E, Brochier G, Labasse C, Madelaine A, Chanut A, Beuvin M, Borsato-Levy F, Biancalana V, Barcia G, De Lonlay P, Laporte J, Böhm J, Romero NB. Acta Neuropathol Commun. 2024 Dec 20;12(1):191. doi: 10.1186/s40478-024-01882-0. PMID: 39707553 Free PMC article.
- Chronic pain as a presenting feature of dysferlinopathy. Sanchez-Casado L, Evangelista T, Nectoux J, Verebi C, Stojkovic T. Neuromuscul Disord. 2025 Jan;46:105269. doi: 10.1016/j.nmd.2024.105269. Epub 2024 Dec 14. PMID: 39798170
- Genomic reanalysis of a pan-European rare-disease resource yields new diagnoses. Laurie S, Steyaert W, de Boer E, Polavarapu K, Schuermans N, Sommer AK, Demidov G, Ellwanger K, Paramonov I, Thomas C, Aretz S, Baets J, Benetti E, Bullich G, Chinnery PF, Clayton-Smith J, Cohen E, Danis D, de Sainte Agathe JM, Denommé-Pichon AS, Diaz-Manera J, Efthymiou S, Faivre L, Fernandez-Callejo M, Freeberg M, Garcia-Pelaez J, Guillot-Noel L, Haack TB, Hanna M, Hengel H, Horvath R, Houlden H, Jackson A, Johansson L, Johari M, Kamsteeg EJ, Kellner M, Kleefstra T, Lacombe D, Lochmüller H, López-Martín E, Macaya A, Marcé-Grau A, Maver A, Morsy H, Muntoni F, Musacchia F, Nelson I, Nigro V, Olimpio C, Oliveira C, Paulasová Schwabová J, Pauly MG, Peterlin B, Peters S, Pfundt R, Piluso G, Piscia D, Posada M, Reich S, Renieri A, Ryba L, Šablauskas K, Savarese M, Schöls L, Schütz L, Steinke-Lange V, Stevanin G, Straub V, Sturm M, Swertz MA, Tartaglia M, Te Paske IBAW, Thompson R, Torella A, Trainor C, Udd B, Van de Vondel L, van de Warrenburg B, van Reeuwijk J, Vandrovcova J, Vitobello A, Vos J, Vyhnálková E, Wijngaard R, Wilke C, William D, Xu J, Yaldiz B, Zalatnai L, Zurek B; Solve-RD DITF-GENTURIS; Solve-RD DITF-ITHACA; Solve-RD DITF-EURO-NMD; Solve-RD DITF-RND; Solve-RD consortium. Nat Med. 2025 Feb;31(2):478-489. doi: 10.1038/s41591-024-03420-w. Epub 2025 Jan 17. PMID: 39825153 Free PMC article.
- High prevalence of facioscapulohumeral muscular dystrophy (FSHD) and inflammatory myopathies association: Is there an interplay? Lauletta A, Allenbach Y, Béhin A, Evangelista T, Léonard-Louis S, Garibaldi M, Benveniste O. J Neurol Sci. 2025 Mar 15;470:123400. doi: 10.1016/j.jns.2025.123400. Epub 2025 Jan 21. PMID: 39855012 Free article.
- Glycogenosis type XI, a rare association between muscle and skin manifestations – the contribution of proteomics for the understanding of the underlying myopathology. Hentschel A, Lacene E, Brochier G, Boisserie JM, Fromes Y, Romero NB, Roos A, Evangelista T. J Neuromuscul Dis. 2025 Mar;12(2):271-278. doi: 10.1177/22143602241296248. Epub 2025 Mar 4. PMID: 40033989 Free article.
- Awareness of bone strength in patients with neuromuscular disorders: ERN EURO-NMD clinician survey and European patient survey. Kruse MTA, Olde Dubbelink BAS, Kroneman M, de Groot I, Schlüter S, de Visser M, Evangelista T, Moretti A, Weber D, Ward LM, Voermans NC; EURO-NMD Bone Strength Study Group. J Neurol Sci. 2025 May 15;472:123420. doi: 10.1016/j.jns.2025.123420. Epub 2025 Feb 7. PMID: 40121805 Free article.
- The most bothersome symptoms in neuromuscular diseases: the ERN EURO NMD Survey. Mancuso M, Colitta A, Lavorato M, Van den Bergh P, Kirschner J, Kornblum C, Maggi L, Lamy F, Lochmüller H, Nordstrøm M, Malfatti E, Ferlini A, Pareyson D, Silani V, Kleopa KA, de Visser M, Atalaia A, Evangelista T. Orphanet J Rare Dis. 2025 May 8;20(1):221. doi: 10.1186/s13023-025-03742-z. PMID: 40340786 Free PMC article.
- Digenic inheritance involving a muscle-specific protein kinase and the giant titin protein causes a skeletal muscle myopathy. Töpf A, Cox D, Zaharieva IT, Di Leo V, Sarparanta J, Jonson PH, Sealy IM, Smolnikov A, White RJ, Vihola A, Savarese M, Merteroglu M, Wali N, Laricchia KM, Venturini C, Vroling B, Stenton SL, Cummings BB, Harris E, Marini-Bettolo C, Diaz-Manera J, Henderson M, Barresi R, Duff J, England EM, Patrick J, Al-Husayni S, Biancalana V, Beggs AH, Bodi I, Bommireddipalli S, Bönnemann CG, Cairns A, Chiew MT, Claeys KG, Cooper ST, Davis MR, Donkervoort S, Erasmus CE, Fassad MR, Genetti CA, Grosmann C, Jungbluth H, Kamsteeg EJ, Lornage X, Löscher WN, Malfatti E, Manzur A, Martí P, Mongini TE, Muelas N, Nishikawa A, O’Donnell-Luria A, Ogonuki N, O’Grady GL, O’Heir E, Paquay S, Phadke R, Pletcher BA, Romero NB, Schouten M, Shah S, Smuts I, Sznajer Y, Tasca G, Taylor RW, Tuite A, Van den Bergh P, VanNoy G, Voermans NC, Wanschitz JV, Wraige E, Yoshimura K, Oates EC, Nakagawa O, Nishino I, Laporte J, Vilchez JJ, MacArthur DG, Sarkozy A, Cordell HJ, Udd B, Busch-Nentwich EM, Muntoni F, Straub V. Nat Genet. 2024 Mar;56(3):395-407. doi: 10.1038/s41588-023-01651-0. Epub 2024 Mar 1. PMID: 38429495 Free PMC article.
- Generation of two iPSC lines from adult central core disease patients with dominant missense variants in the RYR1 gene. Clayton JS, Vo C, Crane J, Scriba CK, Saker S, Larmonier T, Malfatti E, Romero NB, Ravenscroft G, Laing NG, Taylor RL. Stem Cell Res. 2024 Jun;77:103411. doi: 10.1016/j.scr.2024.103411. Epub 2024 Mar 31. PMID: 38582058 Free article.
- Generation of two iPSC lines from patients with inherited central core disease and concurrent malignant hyperthermia caused by dominant missense variants in the RYR1 gene. Clayton JS, Vo C, Crane J, Scriba CK, Saker S, Larmonier T, Malfatti E, Romero NB, Ravenscroft G, Laing NG, Taylor RL. Stem Cell Res. 2024 Jun;77:103410. doi: 10.1016/j.scr.2024.103410. Epub 2024 Mar 30. PMID: 38583293 Free article.
- Exome sequencing in undiagnosed congenital myopathy reveals new genes and refines genes-phenotypes correlations. de Feraudy Y, Vandroux M, Romero NB, Schneider R, Saker S, Boland A, Deleuze JF, Biancalana V, Böhm J, Laporte J. Genome Med. 2024 Jul 9;16(1):87. doi: 10.1186/s13073-024-01353-0. PMID: 38982518 Free PMC article.
- Generation of iPSC lines from three Laing distal myopathy patients with a recurrent MYH7 p.Lys1617del variant. Clayton JS, Vo C, Crane J, Scriba CK, Saker S, Larmonier T, Malfatti E, Romero NB, Ravenscroft G, Laing NG, Taylor RL. Stem Cell Res. 2024 Oct;80:103491. doi: 10.1016/j.scr.2024.103491. Epub 2024 Jul 9. PMID: 39047410 Free article.
- MYH7-related myopathies: clinical, myopathological and genotypic spectrum in a multicentre French cohort. Bahout M, Severa G, Kamoun E, Bouhour F, Pegat A, Toutain A, Lagrange E, Duval F, Tard C, De la Cruz E, Féasson L, Jacquin-Piques A, Richard P, Métay C, Cavalli M, Romero NB, Evangelista T, Sole G, Carlier RY, Laforêt P, Acket B, Behin A, Fernández-Eulate G, Léonard-Louis S, Quijano-Roy S, Pereon Y, Salort-Campana E, Nadaj-Pakleza A, Masingue M, Malfatti E, Stojkovic T, Villar-Quiles RN. J Neurol Neurosurg Psychiatry. 2025 Apr 10;96(5):453-461. doi: 10.1136/jnnp-2024-334263. PMID: 39448255 Free PMC article.
- SH3KBP1 promotes skeletal myofiber formation and functionality through ER/SR architecture integrity. Guiraud A, Couturier N, Christin E, Castellano L, Daura M, Kretz-Remy C, Janin A, Ghasemizadeh A, Del Carmine P, Monteiro L, Rotard L, Sanchez C, Jacquemond V, Burny C, Janczarski S, Durieux AC, Arnould D, Romero NB, Bui MT, Buchman VL, Julien L, Bitoun M, Gache V. EMBO Rep. 2025 Apr;26(8):2166-2191. doi: 10.1038/s44319-025-00413-9. Epub 2025 Mar 10. PMID: 40065183 Free PMC article.
- Disease Trajectories of a Large French Cohort of 142 Congenital Myopathy Patients in Adult Age. Bisciglia M, Severa G, Romero NB, Fardeau M, Rendu J, Stojkovic T, Laforêt P, Eymard B, Ferreiro A, Malfatti E, Béhin A. Eur J Neurol. 2025 Apr;32(4):e70109. doi: 10.1111/ene.70109. PMID: 40159620 Free PMC article.
- Farnè M, Fortunato F, Neri M, Farnè M, Balla C, Albamonte E, Barp A, Armaroli A, Perugini E, Carinci V, Facchini M, Chiarini L, Sansone V, Straudi S, Tugnoli V, Sette E, Sensi M, Bertini M, Evangelista T, Ferlini A, Gualandi F. TeleNEwCARe: An Italian case-control telegenetics study in patients with Hereditary NEuromuscular and CARdiac diseases. EUROPEAN JOURNAL OF MEDICAL GENETICS. 2023;66(6). doi:10.1016/j.ejmg.2023.104749
- Fortunato F, Bianchi F, Ricci G, Torri F, Gualandi F, Neri M, Farne M, Giannini F, Malandrini A, Volpi N, Lopergolo D, Silani V, Ticozzi N, Verde F, Pareyson D, Fenu S, Bonanno S, Nigro V, Peduto C, D’Ambrosio P, Zeuli R, Zanobio M, Picillo E, Servidei S, Primiano G, Sancricca C, Sciacco M, Brusa R, Filosto M, Cotti Piccinelli S, Pegoraro E, Mongini T, Solero L, Gadaleta G, Brusa C, Minetti C, Bruno C, Panicucci C, Sansone VA, Lunetta C, Zanolini A, Toscano A, Pugliese A, Nicocia G, Bertini E, Catteruccia M, Diodato D, Atalaia A, Evangelista T, Siciliano G, Ferlini A. Digital health and Clinical Patient Management System (CPMS) platform utility for data sharing of neuromuscular patients: the Italian EURO-NMD experience. Orphanet J Rare Dis. 2023;18(1):196. doi:10.1186/s13023-023-02776-5
- Lemmers RJLF, Butterfield R, van der Vliet PJ, de Bleecker JL, van der Pol L, Dunn DM, Erasmus CE, D’Hooghe M, Verhoeven K, Balog J, Bigot A, van Engelen B, Statland J, Bugiardini E, van der Stoep N, Evangelista T, Marini-Bettolo C, van den Bergh P, Tawil R, Voermans NC, Vissing J, Weiss RB, van der Maarel SM. Autosomal dominant in cis D4Z4 repeat array duplication alleles in facioscapulohumeral dystrophy. Brain. 2023;147(2):414-426. doi:10.1093/brain/awad312
- Ferreira W, Massaro C, Masingue M, De Lonlay P, Laforet P, Behin A, Eymard B, Choumert A, Mafatti E, Stojkovic T, Allenbach Y, Bassez G, Evangelista T. Rhabdomyolysis and muscle biopsy outcomes: a single center retrospective cohort. NEUROMUSCULAR DISORDERS. 2023;33:S183-S183. doi:10.1016/j.nmd.2023.07.458
- Merlet A, Lacène E, Nelson I, Brochier G, Labasse C, Chanut A, Madelaine A, Beuvin M, Bonne G, Féasson L, Minot M, Noury J, Fradin M, Fernández-Eulate G, Behin A, Stojkovic T, Hentschel A, Marcorelles P, Roos A, Evangelista T. Clinical, morphological, and proteomic features of patients suspected of X-linked myopathy with excessive autophagy (XMEA). NEUROMUSCULAR DISORDERS. 2023;33:S99-S99. doi:10.1016/j.nmd.2023.07.136
- Roos A, van der Ven P, Alrohaif H, Kölbel H, Heil L, Della Marina A, Weis J, Töpf A, Vorgerd M, Schara-Schmidt U, Gangfuss A, Evangelista T, Hentschel A, Grüneboom A, Fuerst D, Kuechler A, Tzschach A, Depienne C, Lochmüller H. Bi-allelic variants of FILIP1 cause congenital myopathy, dysmorphism and neurological defects. NEUROMUSCULAR DISORDERS. 2023;33:S130-S130. doi:10.1016/j.nmd.2023.07.258
- ’t Hoen P, Lalout N, Vroom E, Franken M, Jäger D, Tassoni A, Kampowski T, Delattre H, Hamroun D, Molthof R, de Jong I, Quemada E, Atalaia A, Evangelista T, Wilkinson M, EURO-NMD Registry Consortium. Proven interoperability of five neuromuscular rare disease registries. NEUROMUSCULAR DISORDERS. 2023;33:S145-S145. doi:10.1016/j.nmd.2023.07.312
- Roy B, Peck A, Evangelista T, Pfeffer G, Wang L, Diaz-Manera J, Korb M, Wicklund M, Milone M, Freimer M, Kushlaf H, Villar-Quiles R, Stojkovic T, Needham M, Palmio J, Lloyd T, Keung B, Mozaffar T, Weihl C, Kimonis V. Provisional practice recommendation for the management of myopathy in VCP-associated multisystem proteinopathy. ANNALS OF CLINICAL AND TRANSLATIONAL NEUROLOGY. 2023;10(5):686-695. doi:10.1002/acn3.51760
