

The case of a patient presenting an atypical form of congenital myasthenic syndrome with the identification of a new mutation in the LRP4 gene has just been published in the journal Science Reports*. Result of a close collaboration between French scientists including clinicians from the Service of Neuro-Myology and researchers from the Myology Centre for Research, the article connects the varied clinical manifestations of the disease with the signalling pathways involved at the neuromuscular junction (NMJ) disrupted by the gene mutation. Interview with Dr Marion Masingue**, MD and Stéphanie Bauché***, PhD.
What was the background to this work?
Marion Masingue. In our French cohort of patients with congenital myasthenic syndrome (CMS), we identified a patient who came to the service of Neuro-Myology with Cenani-Lenz syndrome (agenesis of the limbs and a renal malformation), who was very fatigable and therefore suspected of having congenital myopathy.
How did you proceed and what results did you obtain?
MM. We carried out the usual clinical investigations and finally suspected congenital myasthenic syndrome. We then decided to investigate further and carry out more specific tests, in particular JNM, following discussions about this case with colleagues from the SMC**** network. The physiological data obtained, combined with histological data and genetic diagnosis by NGS, enabled us to confirm that this was indeed SMC linked to a new mutation in a known gene, LRP4.
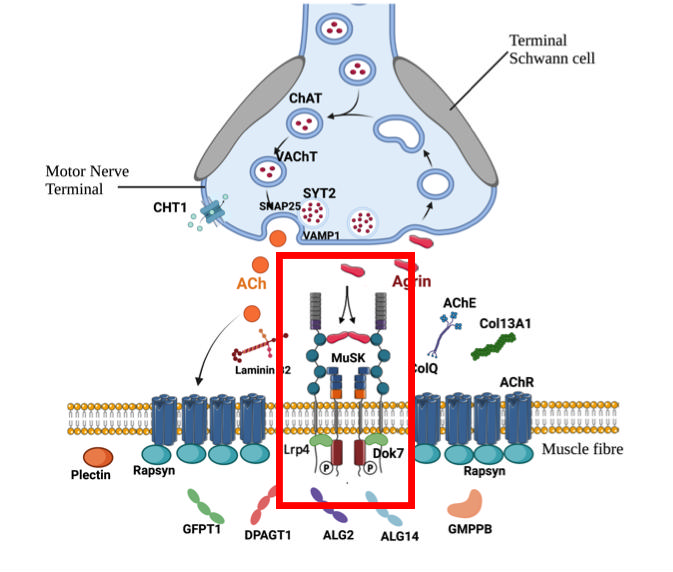
Experiments carried out in vitro, in particular on heterologous cells or muscle cells from the patient’s biopsy, showed a decrease in agrin binding to the mutated domain of LRP4 and a decrease in acetylcholine receptor (AChR) aggregation induced by agrin, resulting in a decrease in the Agrine/LRP4/MuSK signalling pathway, which may explain the decreased neurotransmission (decrement) in the patient associated with her symptoms of muscle weakness and fatigability.
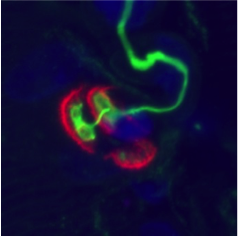
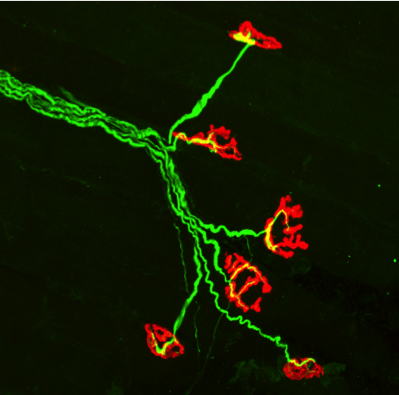 Furthermore, in the same study systems, we showed a decrease in the binding of Wnt11 to the mutated domain, explaining the limb agenesis observed in the patient.
Furthermore, in the same study systems, we showed a decrease in the binding of Wnt11 to the mutated domain, explaining the limb agenesis observed in the patient.
We thus proved that the mutated domain of LRP4 was indeed responsible for the disruption of the agrin/LRP4/MuSK signalling pathway and therefore for the atypical MSC in the patient, but also, from a more fundamental point of view, that this domain of LRP4 was involved in another poorly understood signalling pathway, the Wnt pathway.
Schematic diagram of the NMJ with the Agrine/LRP44/MuSK pathway.
Photographs of immunolabelling of human NMJ.
How can this research improve the lives of patients?
 MM. The originality of this work lies in the fact that we were faced with a very atypical clinical picture in two parts: a MSC with muscular symptoms, and a Cenani-Lenz syndrome with a malformative part. These two conditions had already been described separately but never in association with each other. Exchanges with the SMC network were essential and enabled us to pursue molecular research, to validate the involvement of this new mutation in the patient’s clinical picture, and ultimately to learn more about the Wnt pathway, and therefore more broadly about the pathophysiology of SMC. As the association between malformative symptoms and congenital myasthenia was not known, there are probably a number of people with Cenani-Lenz syndrome who also have SMC and could benefit from targeted treatment. These patients are perhaps a little less rare than we think…
MM. The originality of this work lies in the fact that we were faced with a very atypical clinical picture in two parts: a MSC with muscular symptoms, and a Cenani-Lenz syndrome with a malformative part. These two conditions had already been described separately but never in association with each other. Exchanges with the SMC network were essential and enabled us to pursue molecular research, to validate the involvement of this new mutation in the patient’s clinical picture, and ultimately to learn more about the Wnt pathway, and therefore more broadly about the pathophysiology of SMC. As the association between malformative symptoms and congenital myasthenia was not known, there are probably a number of people with Cenani-Lenz syndrome who also have SMC and could benefit from targeted treatment. These patients are perhaps a little less rare than we think…
SB. In a way, that’s the purpose of all this work: to publish in order to expand global knowledge of MSCs and open up the possibility for all doctors to identify, diagnose and ultimately propose new therapies to improve patients’ lives. Publishing is the only way to communicate with the clinicians who see these patients.
** Marion Masingue is a neurologist practising in the service of Neuro-Myology and at the Reference Centre for Neuromuscular Diseases Nord-Est-Ile de France.
*** Stéphanie Bauché is a research engineer at the Myology Centre for Research, in Antoine Muchir’s team Signaling Pathways & Striated Muscles. The work presented here was carried out when she was part of the Neuromuscular Connectivity in Health & Diseases team led by Bertrand Fontaine and Laure Strochlic.
**** The national Congenital Myasthenic Syndromes network was set up in 2002 and brings together clinicians (neurologists, neuropaediatricians), pathologists, geneticists and neurobiologists who work in synergy to better understand these diseases, identify new mutations and prove their pathogenicity. It is currently led by Arnaud Isapof. Multidisciplinary consultation meetings (RCP) are regularly organised to discuss clinical cases.
