Stéphane Vassilopoulos works in the “Physiopathology & Therapy of Autosomal Dominant Centronuclear Myopathy” laboratory within the team led by Marc Bitoun, at the institute’s Myology Centre for Research. He and his group* are interested in muscle-cell membrane proteins, particularly, clathrin and its implication in myopathies.
What is the link between autosomal dominant centronuclear myopathy and clathrin?
In 2005, Marc Bitoun showed that mutations in the DNM2 gene encoding dynamin 2, were responsible for autosomal dominant centronuclear myopathy (CNM). However, it is known that this protein is essential for clathrin-dependent endocytosis: it forms a spiral which, through its enzymatic action, cleaves the membrane leading to the release of clathrin vesicles. This mechanism is particularly important since cells exchange with the exterior and a large number of therapeutic agents enter the cell via this route.
In 2010, I joined the team to understand the involvement of dynamin through the clathrin pathway, and I began a series of studies on the role of clathrin in muscle cells.
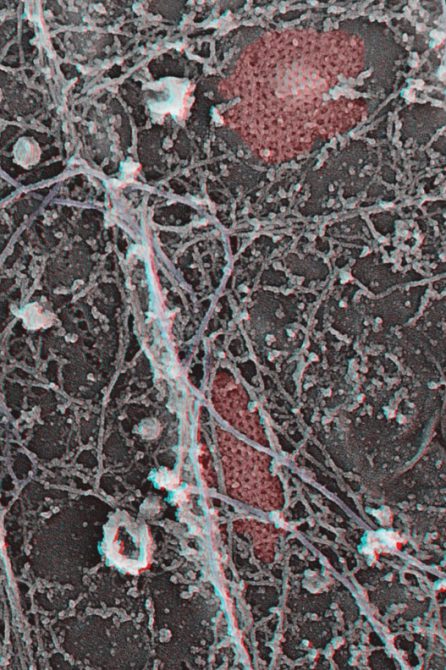
Clathrin is pseudo-coloured in red and the intermediate filaments are purple.
Where did these studies lead you?
For example, the depletion of the clathrin gene by molecular biology techniques leads to an extremely severe and very characteristic muscular dystrophy. We found out by using a platinum replica electron microscopy technique that in cultured cells, the vast majority of clathrin forms flat plaques, networks of hexagons, which are very intimately associated with the cytoskeleton. We have shown that beyond its role in endocytosis, clathrin may play a structural role.
Furthermore, we hypothesised that if the plaques are adhesive structures and that they are associated with the cytoskeleton, then, following mechanical stress such as muscle contractions, they should allow the transmission of mechanical information to the rest of the cell, including the nucleus (ANR-Young Researcher project (2015-2017).
One must understand that the membrane is the Achilles heel of the muscle cell. It is indeed at this level that proteins, which are very often mutated in muscular dystrophies and myopathies, are found: dystrophin and the dystroglycan complex, integrins which connect with the outside of the cell, and many others. However, clathrin organises the membrane, receptor recycling and the cell structure. The defects produced by mutations of the various proteins with which it interacts will therefore generate myopathies such as centronuclear myopathies, but also desminopathies, or myopathies of unknown etiology.
What are the next steps?
We are unique because we combine original electron microscopy and optical microscopy techniques with the current tools of molecular biology. The use of genetically modified systems to specifically disrupt a particular protein makes it possible to precisely determine its role in the organisation of the cell, by visualising what is happening at a very high resolution. We have collaborations with many researchers, and in particular, several teams from the Institute of Myology and in Paris (Curie Institute, Pasteur, Cochin, etc.).
What is important to us is to understand how transitions between endocytosis vesicles (present in all cells) and plaques (only abundant in muscle cells) occur. This should happen through genetic reorganisation. Indeed, most of the proteins that form clathrin plaques are subject to important alternative splicing, and defects in these pathways are involved in the pathophysiology of many myopathies.
*Gilles Moulay, Postdoctoral Fellow, Agathe Frank and Eline Lemerle, PhD students
