L’équipe 5 du Centre de Recherche en Myologie de l’institut « Stratégies thérapeutiques basées sur la réparation de l’ARN & physiopathologie du muscle squelettique » est dirigée par France Pietri-Rouxel. Centrée sur la Dystrophie musculaire de Duchenne, son approche est translationnelle : les travaux qui y sont menés vont de la recherche fondamentale aux essais cliniques. A l’intérieur de l’équipe, quatre groupes thématiques sont portés par quatre chercheurs et composés d’ingénieurs, de personnels techniques et d’étudiants.
Comprendre les anomalies du couplage excitation-contraction
Du côté de la recherche fondamentale, un premier groupe, porté par Sestina Falcone, s’attache à comprendre les anomalies du couplage excitation-contraction. En effet, un muscle ne reste en « bonne santé » que s’il a la capacité de se contracter, c’est un duo entre les motoneurones qui apportent l’activité d’excitation et le muscle qui peut alors se contracter. Lorsqu’il n’y a pas de contraction musculaire le muscle dégénère et s’atrophie. Le couplage excitation-contraction est donc un processus crucial pour le maintien de la masse musculaire et l’étude des anomalies de ce système est primordial dans la compréhension des maladies musculaires (myopathies et notamment Duchenne ou toute autre dystrophie) mais aussi pour le muscle vieillissant. Ce groupe a d’ailleurs identifié des protéines musculaires très importantes qui arrivent à ressentir une anomalie de l’activité électrique et à donner un signal aux muscles pour pouvoir ré-enclencher des mécanismes pour maintenir la masse musculaire et contrecarrer son atrophie. Ces protéines peuvent être envisagées comme des cibles thérapeutiques pour maintenir l’intégrité musculaire et ainsi optimiser les approches de thérapie génique qui ciblent le muscle.
Développer des outils thérapeutiques
Le second groupe de travail, porté par Sofia Benkhelifa-Ziyyat, travaille sur le transport du vecteur AAV dans des vésicules intracellulaires, de la périphérie de la fibre musculaire jusqu’au noyau. En effet, cette étape de transport vésiculaire est cruciale pour la maturation du vecteur, et donc pour une expression efficace du gène thérapeutique dans le noyau de la fibre. Cependant, dans la fibre musculaire Duchenne des anomalies des différents compartiments intracellulaires existent. Le but du projet est d’étudier l’impact de ces anomalies sur le transport du vecteur AAV dans les vésicules intracellulaires et l’expression du transgène dans la fibre dystrophique. A terme, cette étude permettra d’optimiser l’efficacité du traitement avec les vecteurs AAV et de réduire les doses de vecteurs injectés aux patients dans les applications cliniques.
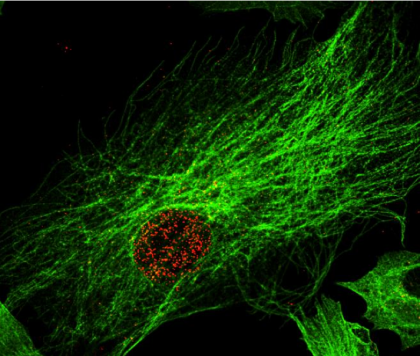
Marquage par immunofluorescence du vecteur AAV dans des cellules musculaires issues de biopsies humaines
Après son entrée dans la cellule musculaire, le vecteur AAV (en rouge) est transporté à travers le réseau microtubulaire (en vert) pour atteindre le noyau cellulaire. C’est dans le noyau de la cellule musculaire que le vecteur AAV va exprimer le gène thérapeutique.
Maintenir les génomes thérapeutiques AAV dans le muscle Duchenne
Un troisième groupe porté par Stéphanie Lorain travaille sur le maintien des génomes thérapeutiques AAV dans le muscle Duchenne. Ce groupe a montré, il y a quelques années, que l’injection de vecteurs AAV est efficace dans un premier temps mais transitoire parce que la fibre dystrophique est fragile et casse, le génome de l’AAV est alors perdu. Or le problème avec la thérapie génique utilisant des vecteurs AAV est que l’on ne peut pas traiter deux fois le patient car il est « vacciné » contre l’AAV par le premier traitement. L’objectif de ce groupe est de trouver un moyen de maintenir le plus possible et le plus longtemps possible le vecteur dans l’organisme pour que le traitement soit pérenne. Dans les derniers travaux publiés en juillet 2016, les chercheurs ont montré qu’en faisant un pré-traitement très fort avec une seule injection de molécules antisens chimiques, les fibres expriment la dystrophine et sont ainsi consolidées pour préparer le muscle à recevoir le traitement AAV. Avec ce pré-traitement, huit fois plus de génomes AAV sont maintenus dans les muscles des souris et dix fois plus de dystrophine est exprimée. Aujourd’hui, un essai pré-clinique avec cette stratégie est envisagé pour traiter toute la musculature d’animaux modèle de la maladie.
Améliorer l’expression de la dystrophine
France Pietri-Rouxel porte le quatrième groupe. Aux portes des essais cliniques, ce groupe a montré, grâce à une étude préclinique, que dans les muscles traités par saut d’exon (AAV-U7), un niveau de restauration de la dystrophine de 40% est nécessaire pour rétablir un état du muscle normal (Gentil et al, 2016). Ce groupe est également impliqué dans l’essai clinique destiné à tester le produit clinique de l’essai AAV-U7-exon 53 prévu dans quelques mois. Ce test est destiné à vérifier que le saut d’exon est efficace et que la dystrophine est restaurée sur les cellules de patients éligibles pour cet essai.
Ce groupe développe également des micro-dystrophines optimisées en évaluant de nombreux critères : la définition des domaines de la protéine qui sont dispensables et indispensables, leur rôle, leur structure et l’impact des nouvelles jonctions lorsqu’un domaine est délété ou ajouté dans une micro-dystrophine. Les chercheurs s’inspirent aussi beaucoup de ce que l’on observe chez les patients atteints de la dystrophie musculaire de Becker qui expriment une dystrophine plus courte mais fonctionnelle. L’objectif est ici de définir la meilleure micro-dystrophine, pour son expression et sa fonction) qui sera apportée par thérapie génique et pourra être une solution thérapeutique pour tous les patients Duchenne.
Le troisième projet de ce groupe concerne une cohorte de patients Becker porteur de la délétion des exons 45 à 55, ces patients présentent une grande hétérogénéité de phénotypes, allant de modéré à sévère. Chez ces patients, la dystrophine, plus courte, a perdu une partie du site de liaison à une protéine associée la nNOS. L’équipe a publié que le phénotype sévère est lié à la perte de liaison du partenaire nNOS (Gentil et al 2012). Ce groupe s’intéresse aujourd’hui à comprendre l’hétérogénéité du phénotype de ces patients en identifiant des modulateurs de sévérité grâce, en particulier, à la technique du whole genome sequencing (séquençage du génome de chaque patient).
